Pathophysiology
It is increasingly being recognized that multiple pathogenetic mechanisms interact in the development of the osteoporotic state. Understanding the pathogenesis of osteoporosis starts with knowing how bone formation and remodeling occur.
Normal bone formation and remodeling
Bone is continually remodeled throughout our lives in response to microtrauma. Bone remodeling occurs at discrete sites within the skeleton and proceeds in an orderly fashion, and bone resorption is always followed by bone formation, a phenomenon referred to as coupling.
Dense cortical bone and spongy trabecular or cancellous bone differ in their architecture but are similar in molecular composition. Both types of bone have an extracellular matrix with mineralized and nonmineralized components. The composition and architecture of the extracellular matrix is what imparts mechanical properties to bone. Bone strength is determined by collagenous proteins (tensile strength) and mineralized osteoid (compressive strength).[18] The greater the concentration of calcium, the greater the compressive strength. In adults, approximately 25% of trabecular bone is resorbed and replaced each year, compared with only 3% of cortical bone.
Osteoclasts, derived from mesenchymal cells, are responsible for bone resorption, whereas osteoblasts, from hematopoietic precursors, are responsible for bone formation (see the images below). The 2 types of cells are dependent on each other for production and linked in the process of bone remodeling. Osteoblasts not only secrete and mineralize osteoid but also appear to control the bone resorption carried out by osteoclasts. Osteocytes, which are terminally differentiated osteoblasts embedded in mineralized bone, direct the timing and location of bone remodeling. In osteoporosis, the coupling mechanism between osteoclasts and osteoblasts is thought to be unable to keep up with the constant microtrauma to trabecular bone. Osteoclasts require weeks to resorb bone, whereas osteoblasts need months to produce new bone. Therefore, any process that increases the rate of bone remodeling results in net bone loss over time.[19]
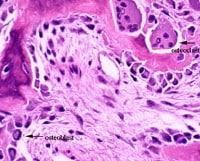 This image depicts bone remodeling with osteoclasts resorbing one side of a bony trabecula and osteoblasts depositing new bone on the other side.
This image depicts bone remodeling with osteoclasts resorbing one side of a bony trabecula and osteoblasts depositing new bone on the other side.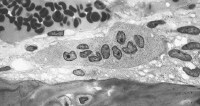 Osteoclast, with bone below it. This image shows typical distinguishing characteristics of an osteoclast: a large cell with multiple nuclei and a "foamy" cytosol.
Osteoclast, with bone below it. This image shows typical distinguishing characteristics of an osteoclast: a large cell with multiple nuclei and a "foamy" cytosol.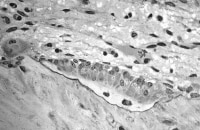 In this image, several osteoblasts display a prominent Golgi apparatus and are actively synthesizing osteoid. Two osteocytes can also be seen.
In this image, several osteoblasts display a prominent Golgi apparatus and are actively synthesizing osteoid. Two osteocytes can also be seen.
Furthermore, in periods of rapid remodeling (eg, after menopause), bone is at an increased risk for fracture because the newly produced bone is less densely mineralized, the resorption sites are temporarily unfilled, and the isomerization and maturation of collagen are impaired.[20]
The receptor activator of nuclear factor-kappa B ligand (RANKL)/receptor activator of nuclear factor-kappa B (RANK)/osteoprotegerin (OPG) system is the final common pathway for bone resorption. Osteoblasts and activated T cells in the bone marrow produce the RANKL cytokine. RANKL binds to RANK expressed by osteoclasts and osteoclast precursors to promote osteoclast differentiation. OPG is a soluble decoy receptor that inhibits RANK-RANKL by binding and sequestering RANKL.
Bone mass peaks around the third decade of life and slowly decreases afterward. A failure to attain optimal bone strength by this point is one factor that contributes to osteoporosis, which explains why some young postmenopausal women have low bone mineral density (BMD) and why some others have osteoporosis. Therefore, nutrition and physical activity are important during growth and development. Nevertheless, hereditary factors play the principal role in determining an individual's peak bone strength. In fact, genetics account for up to 80% of the variance in peak bone mass between individuals.[10, 21]
Alterations in bone formation and resorption
The hallmark of osteoporosis is a reduction in skeletal mass caused by an imbalance between bone resorption and bone formation. Under physiologic conditions, bone formation and resorption are in a fair balance. A change in either—that is, increased bone resorption or decreased bone formation—may result in osteoporosis.
Osteoporosis can be caused both by a failure to build bone and reach peak bone mass as a young adult and by bone loss later in life. Accelerated bone loss can be affected by hormonal status, as occurs in perimenopausal women; can impact elderly men and women; and can be secondary to various disease states and medications.
Aging and loss of gonadal function are the 2 most important factors contributing to the development of osteoporosis. Studies have shown that bone loss in women accelerates rapidly in the first years after menopause. The lack of gonadal hormones is thought to up-regulate osteoclast progenitor cells. Estrogen deficiency leads to increased expression of RANKL by osteoblasts and decreased release of OPG; increased RANKL results in recruitment of higher numbers of preosteoclasts as well as increased activity, vigor, and lifespan of mature osteoclasts.
Estrogen deficiency
Estrogen deficiency not only accelerates bone loss in postmenopausal women but also plays a role in bone loss in men. Estrogen deficiency can lead to excessive bone resorption accompanied by inadequate bone formation. Osteoblasts, osteocytes, and osteoclasts all express estrogen receptors. In addition, estrogen affects bones indirectly through cytokines and local growth factors. The estrogen-replete state may enhance osteoclast apoptosis via increased production of transforming growth factor (TGF)–beta.
In the absence of estrogen, T cells promote osteoclast recruitment, differentiation, and prolonged survival via IL-1, IL-6, and tumor necrosis factor (TNF)–alpha. A murine study, in which either the mice's ovaries were removed or sham operations were performed, found that IL-6 and granulocyte-macrophage CFU levels were much higher in the ovariectomized mice.[22] This finding provided evidence that estrogen inhibits IL-6 secretion and that IL-6 contributes to the recruitment of osteoclasts from the monocyte cell line, thus contributing to osteoporosis.
IL-1 has also been shown to be involved in the production of osteoclasts. The production of IL-1 is increased in bone marrow mononuclear cells from ovariectomized rats. Administering IL-1 receptor antagonist to these animals prevents the late stages of bone loss induced by the loss of ovarian function, but it does not prevent the early stages of bone loss. The increase in the IL-1 in the bone marrow does not appear to be a triggered event but, rather, a result of removal of the inhibitory effect of sex steroids on IL-6 and other genes directly regulated by sex steroids.
T cells also inhibit osteoblast differentiation and activity and cause premature apoptosis of osteoblasts through cytokines such as IL-7. Finally, estrogen deficiency sensitizes bone to the effects of parathyroid hormone (PTH).
Aging
In contrast to postmenopausal bone loss, which is associated with excessive osteoclast activity, the bone loss that accompanies aging is associated with a progressive decline in the supply of osteoblasts in proportion to the demand. This demand is ultimately determined by the frequency with which new multicellular units are created and new cycles of remodeling are initiated.
After the third decade of life, bone resorption exceeds bone formation and leads to osteopenia and, in severe situations, osteoporosis. Women lose 30-40% of their cortical bone and 50% of their trabecular bone over their lifetime, as opposed to men, who lose 15-20% of their cortical bone and 25-30% of trabecular bone.
Calcium deficiency
Calcium, vitamin D, and PTH help maintain bone homeostasis. Insufficient dietary calcium or impaired intestinal absorption of calcium due to aging or disease can lead to secondary hyperparathyroidism. PTH is secreted in response to low serum calcium levels. It increases calcium resorption from bone, decreases renal calcium excretion, and increases renal production of 1,25-dihydroxyvitamin D (1,25[OH]2 D)—an active hormonal form of vitamin D that optimizes calcium and phosphorus absorption, inhibits PTH synthesis, and plays a minor role in bone resorption.
Vitamin D deficiency
Vitamin D deficiency can result in secondary hyperparathyroidism via decreased intestinal calcium absorption.
Osteoporotic fractures
Osteoporotic fractures represent the clinical significance of these derangements in bone. They can result both from low-energy trauma, such as falls from a sitting or standing position, and from high-energy trauma, such as a pedestrian struck in a motor vehicle accident. Fragility fractures, which occur secondary to low-energy trauma, are characteristic of osteoporosis.
Fractures occur when bones fall under excess stress. Nearly all hip fractures are related to falls.[23] The frequency and direction of falls can influence the likelihood and severity of fractures. The risk of falling may be amplified by neuromuscular impairment due to vitamin D deficiency with secondary hyperparathyroidism or corticosteroids.
Vertebral bodies are composed primarily of cancellous bone with interconnected horizontal and vertical trabeculae. Osteoporosis not only reduces bone mass in vertebrae but also decreases interconnectivity in their internal scaffolding.[18]Therefore, minor loads can lead to vertebral compression fractures.
An understanding of the biomechanics of bone provides greater appreciation as to why bone may be susceptible to an increased risk of fracture. When vertical loads are placed on bone, such as tibial and femoral metaphyses and vertebral bodies, a substantial amount of bony strength is derived from the horizontal trabecular cross-bracing system. This system of horizontal cross-bracing trabeculae assists in supporting the vertical elements, thus limiting lateral bowing and fractures that may occur with vertical loading.
Disruption of such trabecular connections is known to occur preferentially in patients with osteoporosis, particularly in postmenopausal women, making females more at risk than males for vertebral compression fractures (see the images below).
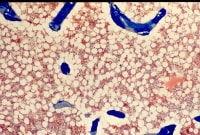 Osteoporosis is defined as a loss of bone mass below the threshold of fracture. This slide (methylmethacrylate embedded and stained with Masson's trichrome) demonstrates the loss of connected trabecular bone.
Osteoporosis is defined as a loss of bone mass below the threshold of fracture. This slide (methylmethacrylate embedded and stained with Masson's trichrome) demonstrates the loss of connected trabecular bone.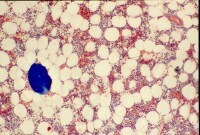 The bone loss of osteoporosis can be severe enough to create separate bone "buttons" with no connection to the surrounding bone. This easily leads to insufficiency fractures.
The bone loss of osteoporosis can be severe enough to create separate bone "buttons" with no connection to the surrounding bone. This easily leads to insufficiency fractures.
Rosen and Tenenhouse studied the unsupported trabeculae and their susceptibility to fracture within each vertebral body and found an extraordinarily high prevalence of trabecular fracture callus sites within vertebral bodies examined at autopsy, typically 200-450 healing or healed fractures per vertebral body.[24] These horizontal trabecular fractures are asymptomatic, and their accumulation reflects the impact of lost trabecular bone and greatly weakens the cancellous structure of the vertebral body.
The reason for preferential osteoclastic severance of horizontal trabeculae is unknown. Some authors have attributed this phenomenon to overaggressive osteoclastic resorption.
Osteoporosis versus osteomalacia
Osteoporosis may be confused with osteomalacia. The normal human skeleton is composed of a mineral component, calcium hydroxyapatite (60%), and organic material, mainly collagen (40%). In osteoporosis, the bones are porous and brittle, whereas in osteomalacia, the bones are soft. This difference in bone consistency is related to the mineral-to-organic material ratio. In osteoporosis, the mineral-to-collagen ratio is within the reference range, whereas in osteomalacia, the proportion of mineral composition is reduced relative to organic material content.
The Wnt signaling pathway and bone
The Wnt family is a highly conserved group of proteins that were initially studied in relationship with cancer initiation and progression due to their involvement in intercellular communication.[25] In the past decade, the Wnt signaling cascade has been recognized as a critical regulator of bone metabolism.
Wnt signaling plays a key role in the fate of mesenchymal stem cells (MSCs), which are the progenitor cells of mature bone-forming osteoblasts.[26] MSCs have the capability to differentiate into adipocytes, chondrocytes, neurons, and muscle cells, as well as into osteoblasts.[27] Certain Wnt signaling pathways promote the differentiation of MSCs along the osteoblast lineage. The emerging details about the specific molecules involved in the Wnt pathway have improved the understanding of bone metabolism and led to the development of new therapeutic targets for metabolic bone diseases.
Wnt signal activation may progress along one of three pathways, with the “canonical” pathway involving β-catenin being most relevant to bone metabolism. The canonical Wnt signaling pathway is initiated by the binding of a Wnt protein to an extracellular co-receptor complex consisting of “Frizzled” (Fr) and low density lipoprotein receptor–related protein–5 or –6 (LRP5, LRP6).[28] This activation recruits another protein, “Disheveled” (Dvl) to the intracellular segment of the Fz/Dvl co-receptor.[29] This is where β-catenin comes into play.
β-Catenin is an important intracellular signaling molecule and normally exists in a phosphorylated state targeted for ubiquination and subsequent degradation within intracellular lysosomes. Activation of the Wnt pathway leads to dephosphorylation and stabilization of intracellular β-catenin and rising cytosolic concentrations of β-catenin. As the concentration of β-catenin reaches a critical level, β-catenin travels to the nucleus, where it activates the transcription of Wnt target genes. Ultimately, canonical Wnt signaling inhibits the expression of transcription factors important in the differentiation of MSCs such as peroxisome proliferator-activated receptor gamma (PPAR-γ) and promotes survival of osteoblast lineage cells.[30]
Several human bone abnormalities have been linked to the Wnt pathway. For example, a single amino acid substitution in the LRP5 receptor gene has been associated with high bone mass phenotypes in humans; specifically, the mutant LRP5 receptor had an impaired interaction with the Wnt signal inhibitor Dickkopf-1 (Dkk-1).[31] Similarly, other missense mutations of LRP5 have been implicated in other high bone mass diseases such as Van Buchem disease and osteopetrosis.[32]Conversely, loss-of-function mutations of LRP5 have resulted in a rare but severe congenital osteoporosis in humans.[33]
There are also several antagonists to the Wnt pathway. Two of the most well-known are Dkk-1 and sclerostin (SOST). Dkk-1 is secreted by MSCs[34] and binds to LRP-5 and LRP-6,[35] thereby competitively inhibiting Wnt signaling. Interestingly, serum levels of Dkk-1 positively correlate with the extent of lytic bone lesions in patients with multiple myeloma.[36] Clinical trials of a monoclonal antibody to Dkk-1 are ongoing.[37]
Similarly, SOST, a product of osteocytes,[38] has also been found to antagonize the Wnt signaling pathway by binding to LRP5 and LRP6.[39] A SOST antibody is also undergoing clinical trials for treatment of metabolic bone disease.[40]
Additional factors and conditions
Endocrinologic conditions or medications that lead to bone loss (eg, glucocorticoids) can cause osteoporosis. Corticosteroids inhibit osteoblast function and enhance osteoblast apoptosis.[41] Polymorphisms of IL-1, IL-6 and TNF-alpha, as well as their receptors, have been found to influence bone mass.
Other factors implicated in the pathogenesis of osteoporosis include polymorphisms in the vitamin D receptor; alterations in insulin-like growth factor-1, bone morphogenic protein, prostaglandin E2, nitrous oxide, and leukotrienes; collagen abnormalities; and leptin-related adrenergic signaling.[19]
Epigenetics
Prenatal and postnatal factors contribute to adult bone mass. In one study, the health of the mother in pregnancy, the infant’s birth weight, and the child’s weight at age 1 year were predictive of adult bone mass in the seventh decade for men and women.[42] It is postulated that growth in the first year of life programs growth hormone that is maintained into the seventh decade.[43] Larger babies and rapid growth in the first year of life predicted increased bone mass in adults aged 65-75 years.
No comments:
Post a Comment